Regenerative Medicine News and General Information
Novel Gene Treatment Can Stop the Growth of Tumors, New Experimental Study Shows
Nov
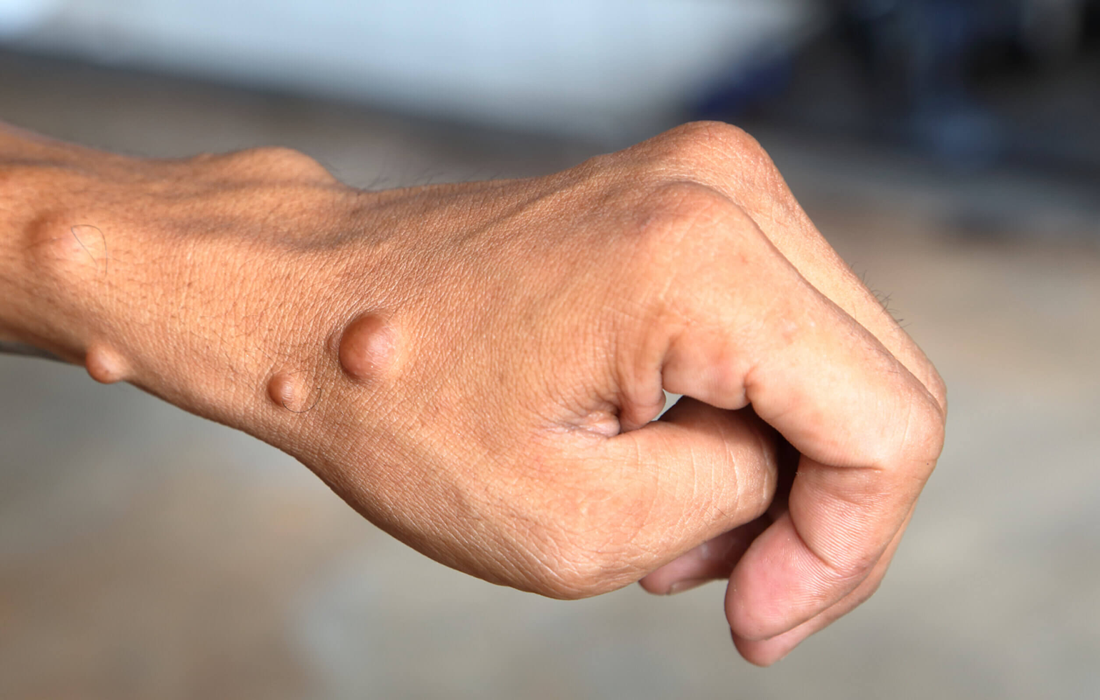
Central neurofibromatosis, or neurofibromatosis type 2 (NF2), is a multisystem genetic disorder associated with bilateral vestibular schwannomas, spinal cord schwannomas, meningiomas, gliomas, and juvenile cataracts, with a paucity of cutaneous features. Schwannomas, most frequently grow on the nerves that bring hearing and balance information into the brain.
Neurofibromatosis type 2 (NF2) is inherited as an autosomal dominant condition, although half of affected individuals have NF2 as a result of a new (de novo) gene mutation. The manifestations of NF2 result from mutations in the NF2 gene, located on the long arm of chromosome 22. Affected individuals need only 1 mutated or deleted NF2 gene to exhibit signs of the condition.
The estimated incidence of neurofibromatosis type 2 (NF2) is 1 in 37,000 per year, with about half of affected individuals representing first cases in the family as a result of new, dominant mutations.
Although the genetic change causing NF2 is present at conception, the clinical manifestations occur over many years. The typical age of onset of symptoms is in the late teens to early 20s, but the age range covers the entire lifespan, to include congenital forms in infancy through the elderly.
Treatment…
Surgical resection of tumors remains the mainstay of treatment in neurofibromatosis type 2 (NF2), with recent advances in surgery permitting preservation of hearing for some affected individuals. For small vestibular schwannomas, surgical resection and stereotactic radiosurgery have been used and may preserve hearing and facial nerve function in selected patients.
Larger tumors may require surgical resection despite irreversible hearing loss, especially when there is evidence of brainstem compression, facial nerve palsy, or, in extreme cases, early hydrocephalus.
Although surgical resection of symptomatic tumors represents the most common approach to clinically significant lesions, in some rare instances, radiation and/or chemotherapy may be recommended to treat disabling ependymomas. However, concerns remain regarding additional risks of radiation therapy in a patient with a germline tumor suppressor gene mutation
What ‘s new?…
An international team of scientists focused on the Hippo signaling pathway, which normally controls organ size in human tissues and cells, but is dysregulated in multiple types of cancer.
Using a combination of patient-donor tumor cells from surgical resections and mouse models of schwannoma, the researchers showed that after just 21 days of the drugs being administered, tumor growth can be strongly and significantly reduced.
In addition, treatment with the Hippo pathway inhibitors (named VT1 and VT2 in the study) actually caused the death of tumor cells and an overall shrinkage of the tumor size.
Initial experiments with these new compounds show they also seem to block the growth of meningioma tumor cells. As well as being a second tumor type seen in patients with NF2, meningioma is overall the most common tumor seen within the brain.
The impact of using these new therapies to hit both tumor types simultaneously has the potential to be highly clinically valuable.
SOURCE:
Liyam Laraba, Lily Hillson, Julio Grimm de Guibert, Amy Hewitt, Maisie R Jaques, Tracy T Tang, Leonard Post, Emanuela Ercolano, Ganesha Rai, Shyh Ming Yang, Daniel J Jagger, Waldemar Woznica, Philip Edwards, Aditya G Shivane, C Oliver Hanemann, David B Parkinson (September 23, 2022). Inhibition of YAP/TAZ-driven TEAD activity prevents growth of NF2-null schwannoma and meningioma. Brain. Retrieved from: https://academic.oup.com/brain/advance-article/doi/10.1093/brain/awac342/6711727
IMAGE:
https://oscestop.education/wp-content/uploads/2022/02/neurofibromatosishand-scaled.jpg